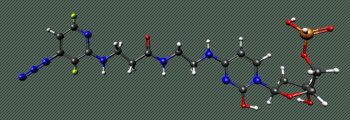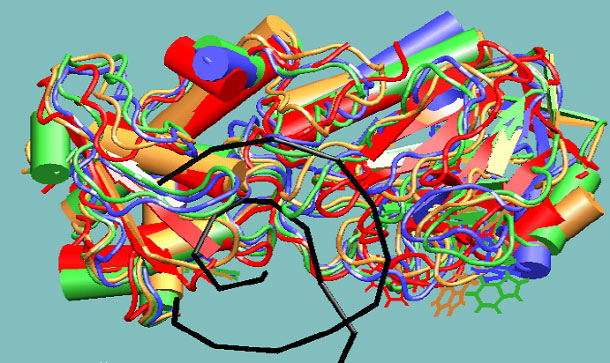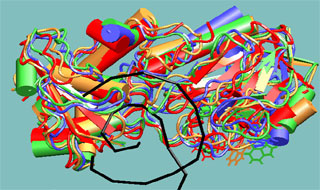
Molecular dynamics study of repair enzyme Fpg bound to a damaged DNA fragment. Red, yellow, green and blue colors correspond to sequential structures generated with molecular dynamics method with 1 ns interval. DNA double helix is displayed in black color. The dynamics of tryptophan aminoacid residues, the actual fluorescent labels, has been studied.
Molecular dynamics (MD) method is a powerful tool to investigate structure of and interaction between macromolecules at an atomic level. MD method enables to render a stochastic thermal fluctuation map of a three-dimensional structure of a molecular system in a water solute, and to study a connection between its submicroscopic texture and biological function; for example, to estimate changes in stability and catalytic properties of enzymes, or a binding affinity of certain medicinal agents or substrate molecules. A practical application of available commercial program complexes indicates that it is difficult to adopt a non-standard biopolymer, that is a biopolymer containing non-canonical aminoacid residues or nucleotides. First, a topology library for aminoacid and nucleotide residues is limited in products distributed. Second, proximity of known force fields and solvation models leaves an opportunity for searching a more precise model that describes biopolymer's energy in water solution. That is why the own original version of modelling program complex for biopolymers using molecular dynamics method, BISON (BIoSimulator Origin Novosibirsk), has been developed.
A pack of five programs comprises the BISON program complex:
- BioPASED (BioPolymer Analysis Structure Energy Dynamics) — implements a molecular dynamics method;
- SIMS — calculation of smooth molecular surface;
- FAMBE — calculation of polarization energy of water solution by solving a Poisson using boundary elements method, and solvation energy;
- FAMBEpH — calculation of protein molecule's energy in water solution for a stated pH accounting for ionization equilibrium and solvation;
- HBDock — implements an exhaustive blind docking of a conformationally mobile ligand and a flexible biopolymer.
Online versions of GUI-BioPASED and GUI-HBDock programs has been developed. The program complex enables: mutant protein structure construction, molecular complex construction, optimization of biopolymer's atomic structure, modelling of thermal fluctuations of the atomic structure using molecular dynamics method, structure validation, construction and docking of ligands. Web versions of programs enable to perform task formation on a remote server as a single executable file via graphical, user-friendly, smart interface, export that file to the client side and ensure its execution. Smart interface allows ordinary users to work with modelling programs, that favours a wider penetration of computer assisted modelling as an auxiliary tool revealing a mechanistic view of biopolymers' interaction for comprehension and planning of molecular biological experiments.